BRUNO EIITI WATABE 1, Henrique Shimidu 2, Leonardo de Paula Ribeiro Figueiredo 3, Thais Fellinger Trindade 4, Victor Dyego Mena Romeiro Nishimura 5
Resumo
A Doença Renal Policística Autossômica Dominante (DRPAD) é uma doença genética comum nos seres humanos sendo caracterizada pelo progressivo crescimento e desenvolvimento de cistos renais bilaterais que culminam com a destruição do parênquima funcional, acarretando em perda função renal e necessidade de diálise na meia idade. Está também associada à manifestações extrarrenais como cistos hepáticos, pancreáticos, aneurismas aórtico e cerebral. Atualmente não existe tratamento específico.Dados do caso
Feminino 59 anos
Palavras chaves
Rim Policístico Autossômico Dominante, Insuficiência Renal Crônica, Doenças Renais Policísticas, Tomografia
Histórico Clínico
Paciente do sexo feminino de 59 anos de idade, comparece ao setor de emergência com quadro de lombalgia bilateral, negava febre e/ou outros sintomas urinários. Como antecedente pessoal referia ser portadora de cistos renais e hepáticos (observados em exame de imagem prévio), hipertensão arterial sistêmica e insuficiência renal crônica. Foi solicitada tomografia computadorizada do abdômen, sem contraste, para pesquisa de nefrolitíase.
Achados Radiológicos
Observam-se rins de dimensões aumentadas, apresentando múltiplos cistos corticomedulares de variados tamanhos. Apesar do tamanho aumentado, há escassez do parênquima renal funcionante, sendo a insuficiência renal crônica uma evolução comum da DRPAD. (Figura 1) Além disso, nota-se o acometimento hepático caracterizado por cistos distribuídos aleatoriamente pelo parênquima do fígado, confluentes na topografia do lobo esquerdo. (Figura 2) Alguns dos cistos renais apresentavam conteúdo hiperdenso que pode representar componente hemorrágico no seu interior, além de calcificações finas das paredes e septos, que podem dificultar a presença de eventuais cálculos renais em exame ultrassonográfico. (Figuras 3 e 4) A tomografia computadorizada evidencia um panorama representativo da doença, sendo um método efetivo na avaliação dos cistos renais, caracterizando de forma satisfatória a sua morfologia, presença ou ausência de septos, sua densidade e eventuais calcificações parietais/septais. (Vídeo 5)
Discussão
A Doença Renal Policística Autossômica Dominante (DRPAD) é causada por mutações nos genes PKD1, correspondendo a 85% dos casos ou PKD2, responsável por 15% , podendo ocorrer mutações "de novo".[1]. Pacientes com mutações em PKD1, particularmente as do tipo "truncating" se apresentam com formas mais severas da doença do que os pacientes com mutações em PKD2. A idade média de apresentação é de 58 e 79 anos, respectivamente. [2] A DRPAD pode se apresentar clinicamente com acometimento renal, extrarrenal e desenvolvimento de comorbidades. Dentre as manifestações extrarrenais podem ser observados cistos hepáticos (em até 44% dos casos) e cistos pancreáticos (10% casos). A doença também está intimamente associada à hipertensão arterial, aneurismas aórticos e cerebrais, além de outras manifestações clínicas como infecção no trato urinário, hematúria , hérnia abdominal, litíase renal e diverticulose intestinal, sendo a dor abdominal e a lombalgia os sintomas mais comuns. [3] A tomografia computadorizada é uma técnica excelente para detectar calcificações além de possibilitar a distinção entre cálculos renais e calcificações dos cistos em pacientes com DRPAD. A ultrassonografia, por sua vez, pode não detectar nefrolitíase nesses pacientes devido a frequente ocorrência de calcificações parietais/septais do cistos renais. [4] Cistos renais associados com DRPAD são corticais e medulares, geralmente simples e tipicamente aumentam em tamanho e número com o tempo. O envolvimento cístico extenso pode ser visto em casos mais severos. Os cistos hepáticos são de origem biliar, sendo comuns em pacientes adultos e raros em pediátricos e geralmente não estão associados a disfunção hepática significativa. [5] A DRPAD é uma doença progressiva que não possui tratamento específico atualmente, o manejo atual consiste em estratégias que incluem o controle pressórico (comumente com medicamentos que interagem com o sistema renina-angiotensina-aldosterona) e minimização da exposição a nefrotoxinas [6]. Cistos dominantes sintomáticos tem sido tratados com aspiração guiada por imagem e escleroterapia com etanol com excelentes resultados a longo prazo [7].
Lista de Diferenciais
Diagnóstico
Aprendizado
A Doença Renal Policística Autossômica Dominante é caracterizada em exames de imagem por múltiplos cistos renais corticomedulares, de tamanhos variados, que aumentam em número e tamanho com o tempo. A presença de calcificações parietais/septais nos cistos e a nefrolitíase, quando concomitantes, podem ser de difícil distinção à ultrassonografia, sendo a tomografia computadorizada uma importante ferramenta diagnóstica nesses casos.
Referências
Imagens
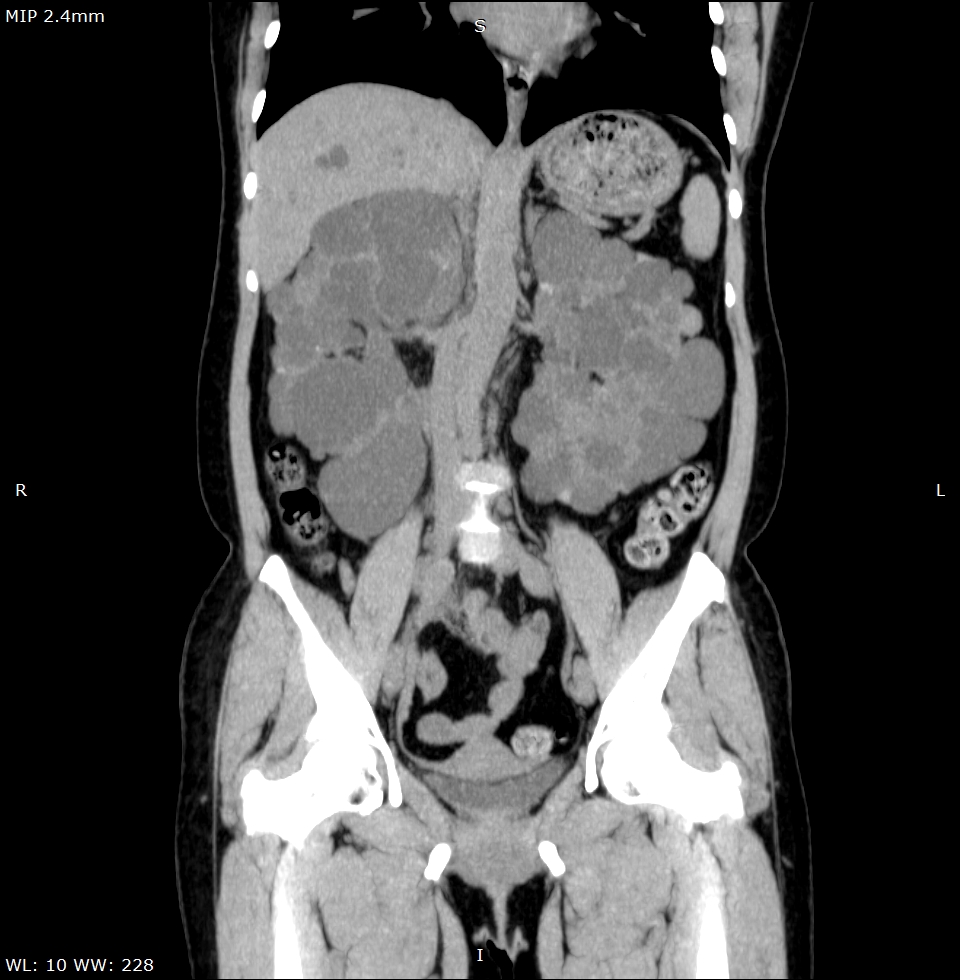
Figura 1. Rins de dimensões aumentadas à custa de múltiplos cistos bilaterais de variados tamanhos.
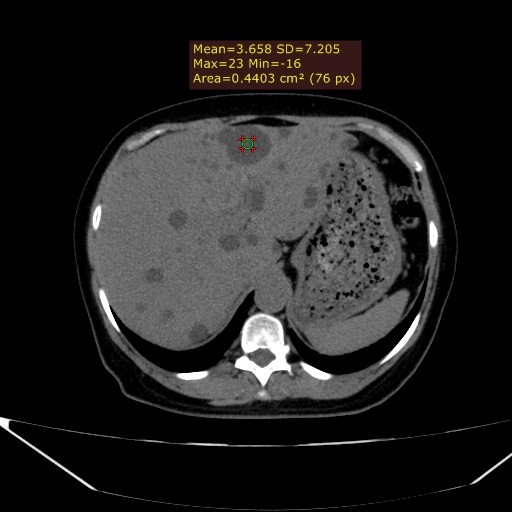
Figura 2. Acometimento hepático com múltiplas lesões de baixa densidade, apresentando aspecto cístico como evidenciado na lesão demarcada pelo ROI.
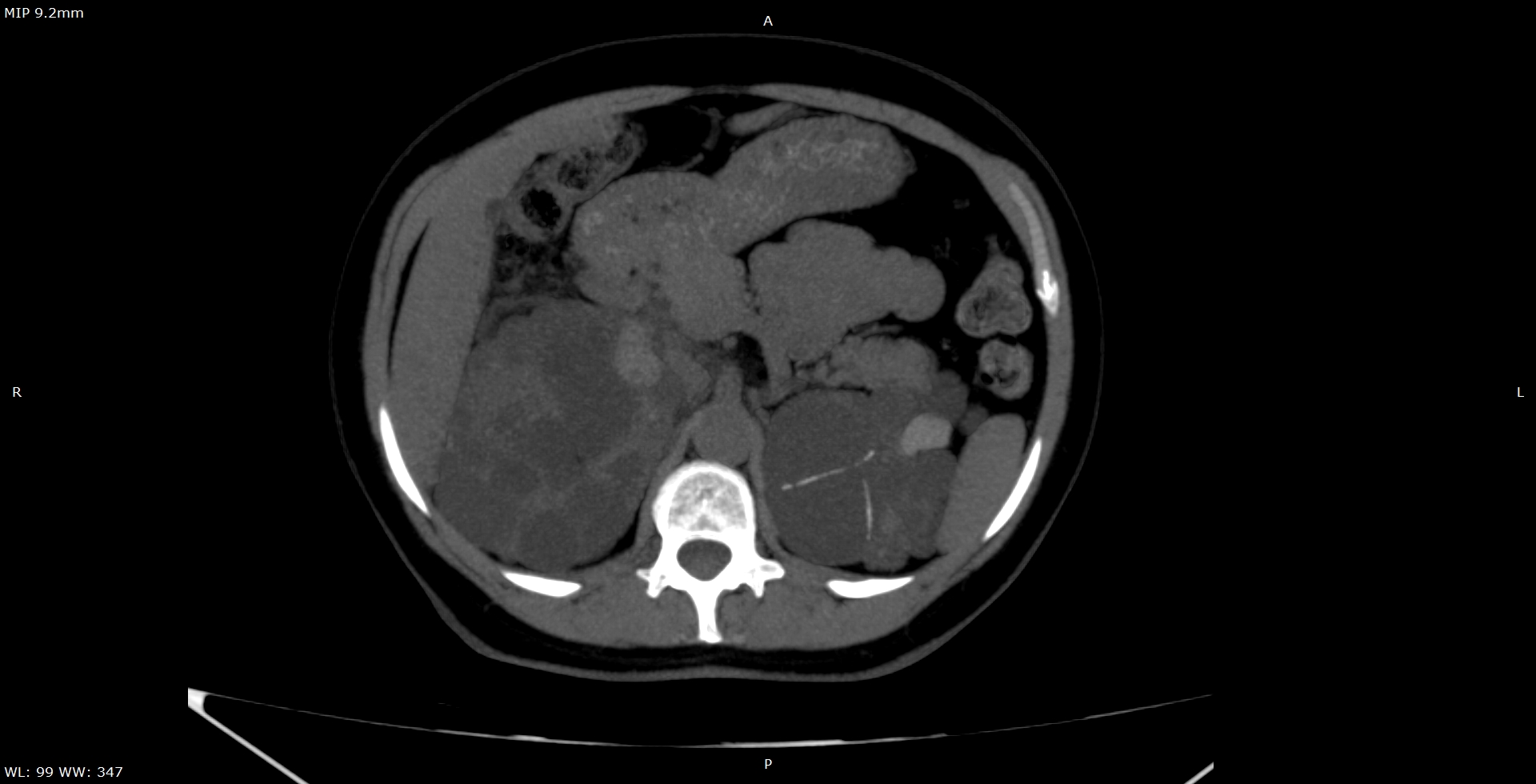
Figura 3. Alguns dos cistos apresentavam finos septos calcificados como observado na figura.
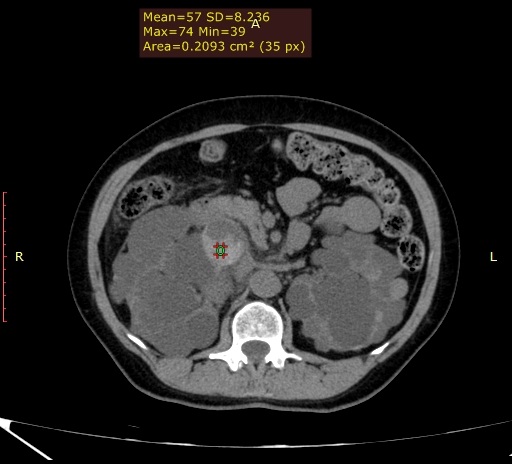
Figura 4. Alguns dos cistos também apresentavam nível líquido-líquido, com componente hiperdenso, apresentando densidade de aproximadamente 57UH como demarcado pelo ROI.
Vídeos
Vídeo 5. Tomografia Computadorizada de abdômen e pelve, realizada sem o uso do meio de contraste iodado por desconhecimento da função renal do paciente no ato do exame (protocolo para pesquisa de nefrolitíase). Nota-se ainda discreto espessamento da fáscia perirrenal à direita, que pode estar relacionada ao rompimento de algum dos cistos.
Artigo recebido em quinta-feira, 9 de julho de 2020
Artigo aprovado em terça-feira, 21 de julho de 2020
 Todos os artigos científicos publicados em brad.org.br são licenciados sob uma licença Creative Commons.
Todos os artigos científicos publicados em brad.org.br são licenciados sob uma licença Creative Commons.
 Português PDF
Português PDF
 Imprimir
Imprimir
 Envie este artigo por e-mail
Envie este artigo por e-mail
 Como citar esse artigo
Como citar esse artigo
 Envie um comentário
Envie um comentário
 Mendeley
Mendeley
 Pocket
Pocket