Maria Luiza de Mello Pereira 1, Antonio Soares Souza 2, Mariana Ribeiro Rodero Cardoso 3, Isis Forgerini 4
Resumo
A Síndrome de Sturge-Weber (SSW) ou angiomatose encefalotrigeminal, é uma facomatose rara, caracterizada por malformações vasculares dérmicas da face, das leptomeninges e olhos. Sintomas extracutâneos incluem convulsões, atraso no desenvolvimento neuropscicomotor e glaucoma. Descrevemos aqui um caso de SSW com acometimento encefálico bilateral, cuja investigação foi iniciada devido à síndrome epiléptica de difícil controle acompanhada de hemangiomas generalizados na face e dorso.Dados do caso
Masculino, 3 anos.
Palavras chaves
Epilepsia, Síndrome de Sturge-Weber, Hemangioma, Glaucoma, Deficiências do Desenvolvimento.
Histórico Clínico
Paciente do sexo masculino, 3 anos e 9 meses, encaminhado ao serviço do Hospital da Criança e Maternidade de São José do Rio Preto, para investigação de atraso no desenvolvimento neuropsicomotor, epilepsia de difícil controle, hemangiomas generalizados na face e dorso, perda severa da audição, glaucoma congênito bilateral e laringotraqueomalácia.
Achados Radiológicos
O estudo por ressonância magnética é o mais específico para o diagnóstico da SWW, demonstrando marcada redução volumétrica (Figura 2) e angiomatose pial com realce leptomeníngeo (Figura 1), isquemias venosas congestivas, e dilatação de vias transparenquimatosas por drenagem venosa profunda anormal. [3] O plexo coróide é frequentemente aumentado de acordo com a extensão do angioma leptomeníngeo. Em T2, nota-se baixo sinal da substância branca subjacente aos angiomas, e em GE/SWI/ EPI, evidencia-se queda de sinal por calcificações distróficas grosseiras giriformes no córtex cerebral. [2] Há espessamento da calvária em associação com a atrofia cerebral, alargamento compensatório dos seios paranasais e de células da mastoide. A tomografia computadorizada é mais sensível na detecção de calcificações corticais e subcorticais (Figura 3). [2,3]
Discussão
A SSW, também denominada angiomatose encefalotrigeminal, é uma doença congênita neurocutânea rara, caracterizada por angiomas envolvendo as leptomeninges, a coroide do olho e a pele da face, geralmente nas regiões inervadas pelos ramos oftálmico e maxilar do nervo trigêmeo. Podem também ocorrer angiomas extracranianos e de partes moles. [1,2] Esta facomatose faz parte de um amplo espectro de fenótipos incluídos na síndrome arteriovenosa metamérica craniofacial (CAMS) e é causada por persistência de vasos do plexo transitório sinusoidal primordial, resultando nas malformações supracitadas. Além da malformação vascular meníngea, evidencia-se com frequência atrofia subjacente do hemisfério cerebral, sendo o acometimento unilateral mais comum. A maioria dos pacientes (80%) têm epilepsia de difícil controle levando a rápida deterioração neurológica, e mais de 50% têm deficiência mental. [2,4] Os exames imaginológicos têm relevância diante da suspeição clínica e dentre os principais métodos utilizados estão a tomografia computadorizada (TC) e a ressonância magnética (RM). A TC é mais sensível que a RM na detecção de calcificações corticais e subcorticais, porém, a RM tem alta sensibilidade e especificidade na detecção de alterações na mielinização, além de demonstrar com nitidez exuberante realce leptomeníngeo. Ainda, permite boa representação de malformações orbitais associadas à síndrome. [2,3] Cerca de 30% dos pacientes têm angiomas de coroide e esclera. A presença de angioma de coroide se correlaciona com a presença de doença bilateral, entretanto, a extensão do envolvimento facial não tem associação com o tamanho do angioma pial intracraniano. [2] Contudo, outras causas de calcificação intracraniana e padrões angiográficos semelhantes como os encontrados nas síndromes de Klippel-Trenaunay-Weber e de Wyburn-Mason, podem não ser diferenciados daqueles evidenciados na SSW.
Lista de Diferenciais
Diagnóstico
Aprendizado
Apesar do diagnóstico da síndrome ser clínico, o estudo imaginológico tem papel fundamental na avaliação da extensão das lesões e gravidade da anormalidade, resultando em disfunção neuronal e outros sintomas neurológicos. O acometimento bilateral como o apresentado aqui é bastante raro, reforçando a importância do seu relato e auxiliando no diagnóstico por radiologistas menos experientes. Os achados de imagem podem ainda ser úteis no manejo e implicações prognósticas.
Referências
1 Takeoka M, Riviello Jr JJ. Sturge-Weber Syndrome. Medscape. 05 January 2010 [cited 2011 Jan 11].
2 Adroos N, Smal J, Suleman FE. Double trouble: Bilateral cerebral involvement in SturgeWeber syndrome. S Afr J Rad. 2015;19(1); Art. #760, 4 pages. ].
3 Khan NA, et al Imaging in Sturge-Weber Syndrome. Medscape. 20 November 2015.
4 Cafe, MEM; Rodrigues, RC; Viggiano, AM. Você conhece esta síndrome?. An. Bras. Dermatol., Rio de Janeiro, v. 83, n. 2, p. 167-169, Apr. 2008.
5 Cagneaux M, Paoli V, Blanchard G, Ville D, Guibaud L. Pre- and postnatal imaging of early cerebral damage in Sturge-Weber syndrome. PediatrRadiol. 2013 Nov. 43 (11):1536-9. [Medline].
Imagens
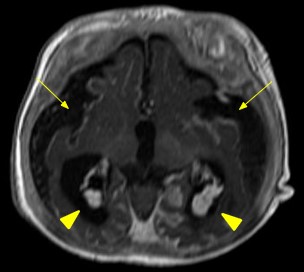
Figura 1: Imagem axial de RM ponderada em T1 pós-gadolínio demonstrando realce pial (setas finas) e impregnação difusa, além de engurgitamento dos plexos coroides (cabeças de seta)
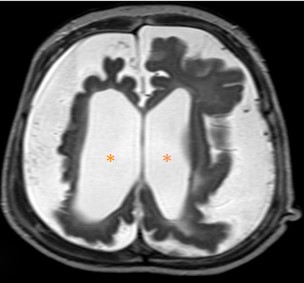
Figura 2: Corte axial de RM encefálica ponderada em T2, evidenciando redução volumétrica encefálica difusa, configurando atrofia e dilatação compensatória do sistema ventricular supratentorial (*)
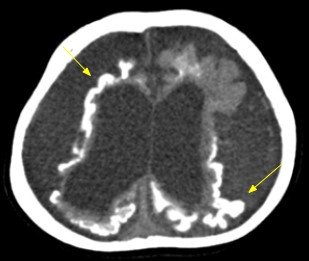
Figura 3: Imagem axial de tomografia computadorizada sem uso de contraste endovenoso demonstrando acentuação severa e difusa dos sulcos entre os giros corticais com dilatação compensatória assimétrica dos ventrículos laterais. Também, calcificações grosseiras giriformes no córtex cerebral (setas finas)
Artigo recebido em quarta-feira, 26 de janeiro de 2022
 Todos os artigos científicos publicados em brad.org.br são licenciados sob uma licença Creative Commons.
Todos os artigos científicos publicados em brad.org.br são licenciados sob uma licença Creative Commons.
 Português PDF
Português PDF
 Imprimir
Imprimir
 Envie este artigo por e-mail
Envie este artigo por e-mail
 Como citar esse artigo
Como citar esse artigo
 Envie um comentário
Envie um comentário
 Mendeley
Mendeley
 Pocket
Pocket