Marina Shu Fong 1
Resumo
A síndrome da persistência do ducto Mülleriano é uma forma rara de pseudo-hermafroditismo em homens, causada pela deficiência na liberação intra-uterina de hormônio anti-Mülleriano (HAM), bem como defeitos no seu receptor. Nesta síndrome congênita, um homem com genótipo 46XY e fenótipo normal apresenta órgãos genitais femininos (útero, trompas de falópio e terço superior da vagina) derivados dos ductos de Müller na vida fetal.Dados do caso
Masculino, 39 anos.
Palavras chaves
Hormônio Antimülleriano, Transtornos do Desenvolvimento Sexual.
Histórico Clínico
Paciente masculino em investigação de infertilidade, com história prévia de orquidectomia esquerda para testículo não descido. No exame clínico, os testículos não eram palpáveis no saco escrotal. Desenvolvimento normal do pênis e uretra, bem como dos caracteres sexuais secundários, como bigode, barba, pêlos pubianos e axilares. Os detalhes da cirurgia não estavam disponíveis.
Achados Radiológicos
A ressonância magnética mostrou estrutura pélvica com morfologia semelhante ao útero e colo uterino (figura 1), com três zonas definidas e terço superior da vagina em fundo cego, posterior à bexiga, em contiguidade com a superfície posterior da próstata e à vesícula seminal esquerda hipoplásica (figuras 2 e 3). Havia mínima quantidade de líquido no interior da cavidade endometrial. Os testículos, ductos deferentes, ductos ejaculatórios, epididímos e vesícula seminal direita não foram vizibilizados. Nenhuma estrutura consistente com a morfologia ovariana foi observada. A próstata (figura 4) e o pênis (figura 5) apresentavam tamanho e morfologia normais. Alterações pós cirúrgicas relacionadas a orquidectomia esquerda para testículo não descido, eram observadas na região inguinal esquerda (figura 6).
Discussão
A síndrome da persistência do ducto Mülleriano (SPDM) representa uma forma rara de pseudo-hermafroditismo em homens, com transmissão autossômica recessiva e poucos casos com possível herança ligada ao X, causada pela deficiência na liberação intra-uterina de hormônio anti-Mülleriano (HAM), bem como defeitos no seu receptor. Nesta síndrome congênita, um homem com genótipo 46XY e fenótipo normal apresenta órgãos genitais femininos (útero, trompas de falópio e terço superior da vagina) derivados do ducto de Müller na vida fetal. Em um desenvolvimento fetal masculino habitual, os ductos de Müller e Wolff estão ambos presentes na sétima semana de gestação. O ducto de Wolff se desenvolverá normalmente, formando os órgãos reprodutores masculinos; e posteriormente, o ducto de Müller irá regredir a partir da ação do HAM produzido pelas células de Sertoli do feto masculino. A testosterona produzida pelos testículos fetais apresenta efeito local direto nos ductos wolffianos, incluindo a diferenciação em epidídimos, ducto deferente e vesículas seminais, e a formação do seio urogenital e da genitália externa masculina requer conversão in situ da testosterona em di-hidrotestosterona. No paciente com a SPDM, o HAM não é liberado; portanto, o feto irá formar órgãos reprodutores masculinos e femininos derivados do ducto de Wolff e Müller, respectivamente. Existem tem 2 tipos anatômicos na SPDM: masculino e feminino. O tipo masculino é mais comum (em até 90% dos casos), classificado em duas subcategorias: hérnia uterina inguinal (em sua maioria com criptorquidia unilateral e hérnia inguinal contralateral - figura), e ectopia testicular transversa (rara, com herniação de testículos e estruturas Müllerianas no mesmo saco herniário na região inguinal). A variante feminina da SPDM é ainda menos comum (10-20%), caracterizada pela presença de útero fixo na pelve e ambos os testículos em meio aos ligamentos redondos. Muitos casos de SPDM podem ser difíceis de diagnosticar ou ainda subdiagnosticados devido à raridade da entidade, sendo que existem menos de 300 casos na literatura, diagnosticados predominantemente em pacientes com menos de 10 anos de idade. O diagnóstico é frequentemente incidental durante o reparo de hérnia inguinal ou em cirurgias para testículos não descidos. A história clínica e estudo genético auxiliam no diagnóstico, principalmente na distinção com outros distúrbios intersexuais. Cariótipo e avaliação da resposta testicular à estimulação da gonadotrofina coriônica são essenciais para verificar o sexo genético e a existência de tecido testicular funcional.
Lista de Diferenciais
Diagnóstico
Aprendizado
A SPDM é uma condição rara, diagnosticada incidentalmente durante o tratamento cirúrgico de hérnia inguinal ou testículo não descido, cujo principal método diagnóstico é através da imagem pré-operatória, como por ressonância magnética. É de extrema importância nos familiarizarmos com esse distúrbio incomum e suas formas de apresentação distintas, para um adequado planejamento pré-operatório e prevenção de complicações futuras, como infertilidade masculina e transformação maligna testicular.
Referências
1. Alharbi K N. Radiological Findings in Persistent Müllerian Duct Syndrome: Case Report and Review of Literature. J Radiol Case Rep. 2017 Mar; 11(3): 7–14.
3. Dekker H. Best Cases from the AFIP. RadioGraphics. Mar 1 2003; 23 (2).
4. Aw L D. Persistent Mullerian Duct Syndrome: a rare entity with a rare presentation in need of multidisciplinary management. International braz j urol, 2016. 42(6), 1237-1243.
2. Singh R. MRI findings of Persistent Mullerian Duct Syndrome: A Rare Case Report. J Clin Diagn Res. 2017 Jun; 11(6): TD05–TD06.
5. Picard J. The Persistent Müllerian Duct Syndrome: An Update Based Upon a Personal Experience of 157 Cases. Sex devel journ 11 3 (2017): 109-125 .11 3 (2017): 109-125.
6. Ferreira DM et al. Magnetic resonance imaging of the vagina Radiol Bras. 2015 Jul/Ago;48(4):249–259 249 0100-3984
Imagens
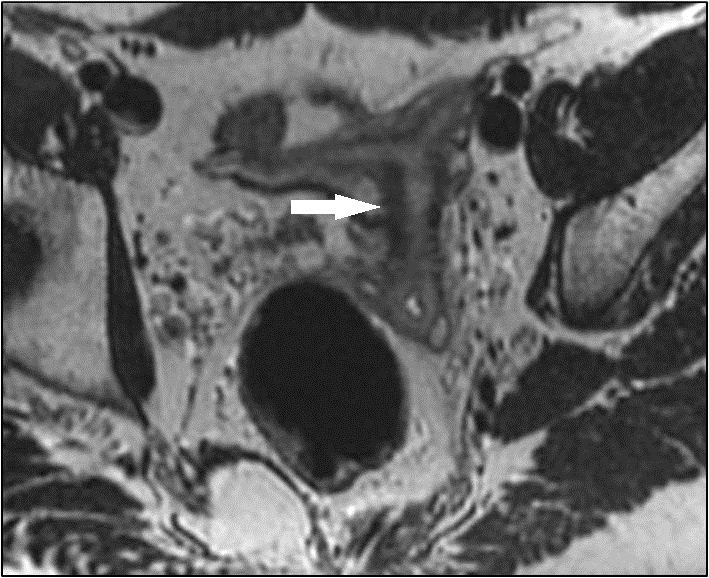
Figura 1 - RM coronal T2 mostra estrutura alongada pélvica à esquerda, com morfologia semelhante ao útero e distensão da cavidade endometrial (→).
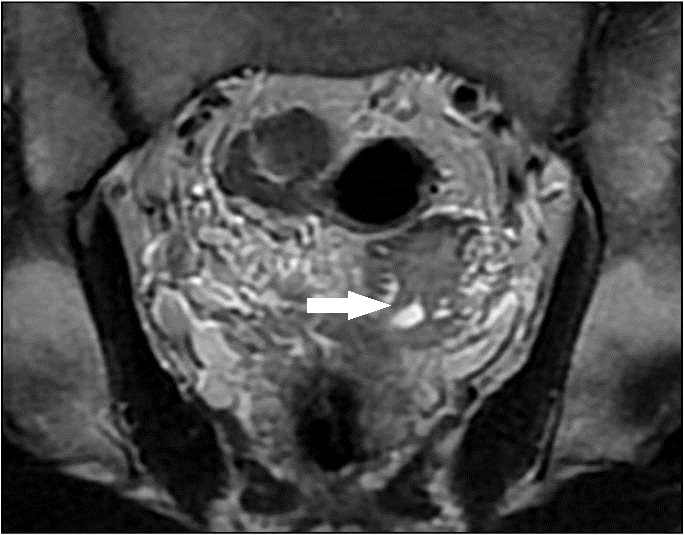
Figuras 2 e 3 - RM coronal T2 mostra que a estrutura alongada pélvica à esquerda, com morfologia semelhante ao útero e distensão da cavidade endometrial, apresentava contiguidade mostra vesícula seminal esquerda hipoplásica (→) junto à base prostática (*).
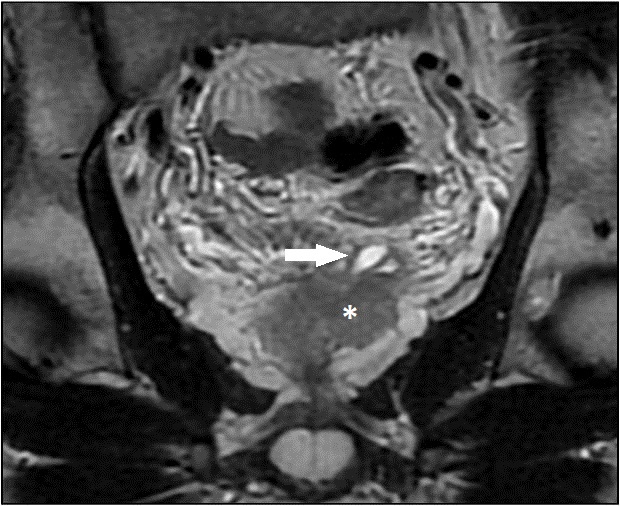
Figuras 2 e 3 - RM coronal T2 mostra que a estrutura alongada pélvica à esquerda, com morfologia semelhante ao útero e distensão da cavidade endometrial, apresentava contiguidade mostra vesícula seminal esquerda hipoplásica (→) junto à base prostática (*).
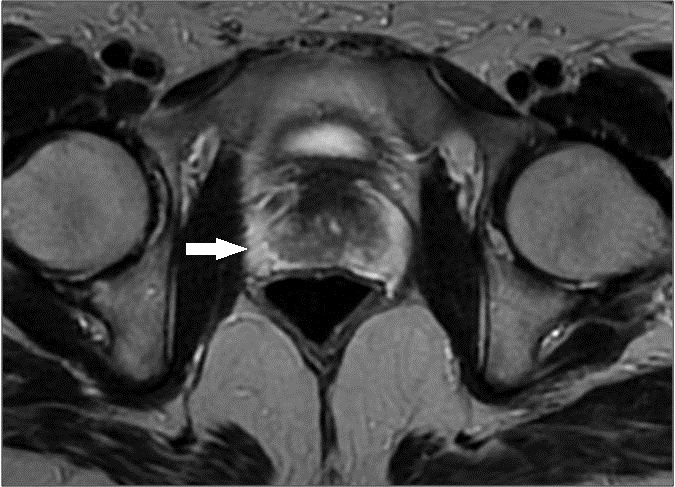
Figura 4 - RM axial T2 próstata com aspecto morfológico normal (→).
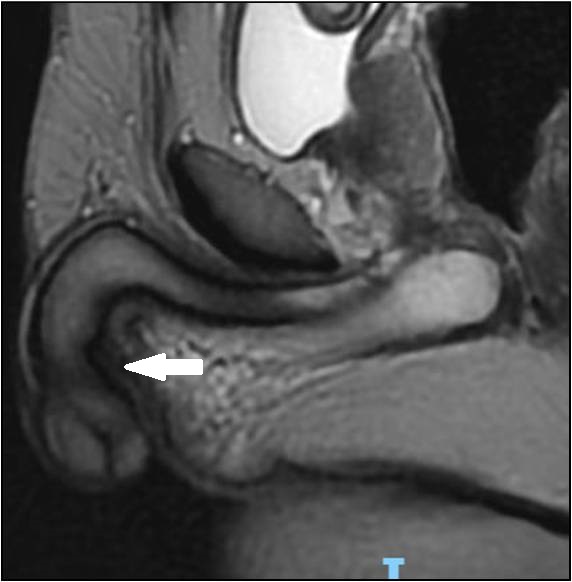
Figura 5 - RM sagital T2 pênis com aspecto morfológico habitual (→).
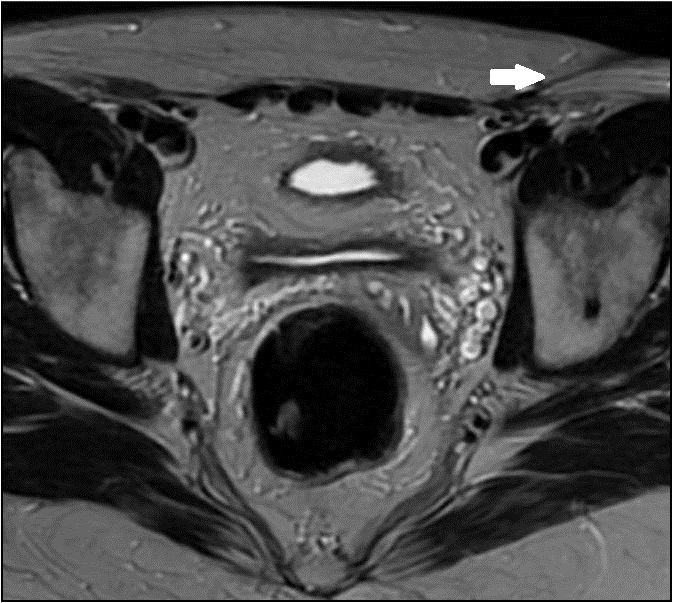
Figura 6 - RM axial T2 alterações pós cirúrgicas na região inguinal esquerda, relacionadas à orquiectomia ipsilateral (→).
Artigo recebido em sexta-feira, 20 de março de 2020
Artigo aprovado em segunda-feira, 30 de março de 2020
 Todos os artigos científicos publicados em brad.org.br são licenciados sob uma licença Creative Commons.
Todos os artigos científicos publicados em brad.org.br são licenciados sob uma licença Creative Commons.
 Português PDF
Português PDF
 Imprimir
Imprimir
 Envie este artigo por e-mail
Envie este artigo por e-mail
 Como citar esse artigo
Como citar esse artigo
 Envie um comentário
Envie um comentário
 Mendeley
Mendeley
 Pocket
Pocket