João Antonio Pessôa Corrêa 1, Maria Lucia Borri 2
Abstracto
Doenças priônicas são um grupo heterogêneo de doenças neurodegenerativas raras, sendo a Doença de Creutzfeldt-Jakob a mais comum delas, notadamente sua forma esporádica. Esta ocorre por acúmulo da proteína insolúvel PrPsc (proteínas priônicas alteradas). É um importante diagnóstico diferencial de demência rapidamente progressiva, e a ressonância magnética apresenta padrões típicos que auxiliam na suspeita, uma vez que o diagnóstico definitivo só é possível por meio do estudo histopatológico.Dados do caso
Masculino, 53 anos.
Palavras chaves
Doenças Priônicas, Síndrome de Creutzfeldt-Jakob, Imagem por Ressonância Magnética.
Histórico Clínico
Paciente do sexo masculino, 53 anos, apresentando diplopia, tontura, fraqueza nos membros inferiores e perda ponderal de início há dois meses, com surgimento de fasciculações e alucinações visuais e persecutórias há um mês e desequilíbrio acentuado levando a incapacidade de deambular, tremor, disfagia e incontinência urinária e fecal anterior à internação. Ao exame físico, apresentava força grau II nos membros inferiores e III nos superiores, disdiadococinesia, dismetria e fasciculação lingual. Durante a investigação, foram realizados ressonância magnética e análise do líquido cefalorraquidiano, demonstrando achados sugestivos de doença de Creutzfeldt-Jakob relatados a seguir.
Achados Radiológicos
Ressonância magnética demonstra sinal hiperintenso na sequência FLAIR associado a restrição à difusão envolvendo difusamente o córtex, assimétrico e predominando no hemisfério cerebral esquerdo, e núcleos caudados e putâmens. As imagens pesadas em T2 não apresentam alterações evidentes.
Discussão
A doença de Creutzfeldt-Jakob (DCJ) esporádica é uma patologia rara, sendo doença priônica mais comum em humanos. [1,2] É rapidamente progressiva e invariavelmente fatal. [3] Ocorre por conversão esporádica de proteína priônica existente naturalmente (PrPc) em proteína priônica anormal (PrPsc). [2] São achados histopatológicos a microvacuolização degenerativa da substância cinzenta (degeneração espongiforme) com perda neuronal, gliose e acúmulo de PrPsc. [1,2] O Center for Disease Control (CDC) norte-americano preconiza que o diagnóstico definitivo da DCJ esporádica é histopatológico. [3] Sendo assim, é rara a realização de diagnóstico definitivo, por ser invasivo. O diagnóstico provável pode ser feito associando-se achados clínicos a ao menos um teste paraclínico. Os achados clínicos são a demência rapidamente progressiva associada a ao menos dois dos seguintes sintomas: mioclonia, sinais visuais ou cerebelares, sinais piramidais/extrapiramidais e/ou mutismo acinético. São considerados testes paraclínicos, com achados típicos, o eletroencefalograma (complexos periódicos de ponta-onda), análise do líquido cefalorraquidiano (presença de proteína 14-3-3) e ressonância magnética (RM). Diagnósticos alternativos devem ser excluídos. [3] Na RM, os achados típicos descritos são hipersinal na sequência FLAIR e, principalmente, em DWI, de distribuição cortical e nos núcleos da base. A causa da restrição à difusão ainda não é clara, podendo ser atribuída à restrição da difusibilidade da água pela compartimentalização nos vacúolos ou pela deposição de PrPsc. [1] No córtex, a alteração de sinal pode ser focal ou difusa, simétrica ou assimétrica e em geral poupa a região perirrolândica. [1,2] Nos núcleos da base, os achados podem ser simétricos ou assimétricos, acometendo principalmente os núcleos caudados e putâmens, além de apresentar um gradiente antero-posterior. A alteração de sinal nos núcleos da base apresenta aumento em extensão e intensidade de sinal a medida que a doença progride. Atrofia do cerebelo é descrita na DCJ esporádica. [1] Os achados radiológicos típicos podem preceder as manifestações clínicas. [1] O caso em discussão apresenta alterações típicas da DCJ esporádica na RM, podendo ser utilizado como um critério de diagnóstico provável. (Figs. 1-4) Algumas alterações atípicas podem ser encontradas, como hipersinal em T2, FLAIR e DWI na região posterior (pulvinar) e dorsomedial do tálamo (sinais do "pulvinar" e do "taco de hockey"), mais característico na forma variante da DCJ, hipersinal em T1 no globo pálido e hipersinal no DWI do cerebelo. [1]
Lista de Diferenciais
Diagnóstico
Aprendizado
Apesar do diagnóstico definitivo ser histopatológico, a clínica unida a achados paraclínicos, incluindo a ressonância magnética, determinam o diagnóstico provável da doença. É necessário o conhecimento dos padrões de imagem pelo radiologista, a fim de auxiliar no diagnóstico não invasivo da doença de Creutzfeldt-Jakob esporádica.
Referências
1. Fragoso D, Gonçalves Filho A, Pacheco F, Barros B, Aguiar Littig I, Nunes R et al. Imaging of Creutzfeldt-Jakob Disease: Imaging Patterns and Their Differential Diagnosis. RadioGraphics. 2017;37(1):234-257.
2. Koeller K, Shih R. Viral and Prion Infections of the Central Nervous System: Radiologic-Pathologic Correlation:From the Radiologic Pathology Archives. RadioGraphics. 2017;37(1):199-233.
3. Creutzfeldt-Jakob disease, classic (CJD). Centers for Disease Control and Prevention website. Acessado em 29/04/2020. Disponível em: http://www.cdc.gov/prions/cjd/index.html. Atualizado em 9 de outubro de 2018.
Imagens
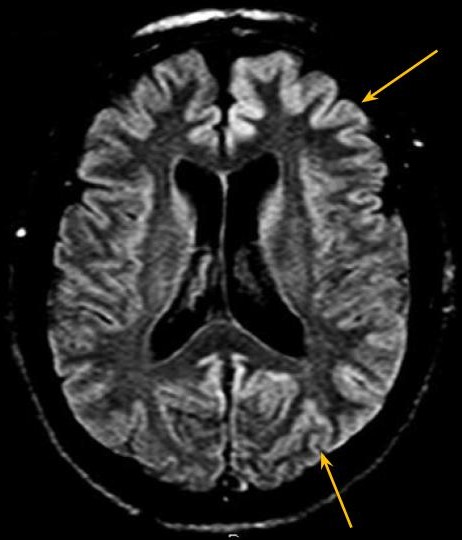
Fig.1 - Sequência FLAIR, plano axial, demonstrando hipersinal cortical assimétrico, predominando nos giros frontais e occipitais à esquerda.
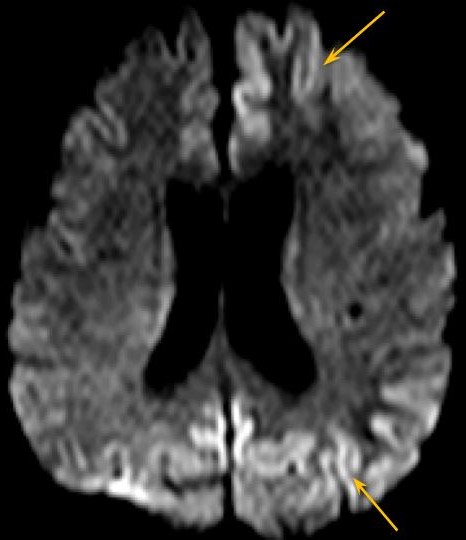
Fig.2 - Sequência DWI, plano axial, apresentando restrição a difusão nas mesmas regiões corticais com alteração no FLAIR
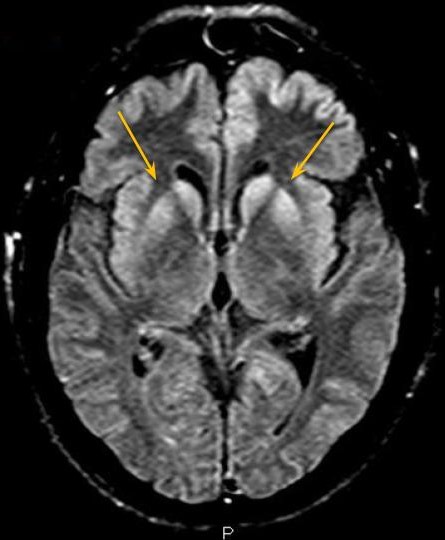
Fig.3 - Sequência FLAIR, plano axial mais inferior, observando-se hipersinal nos núcleos caudados e putâmens, além do hipersinal cortical observado previamente.
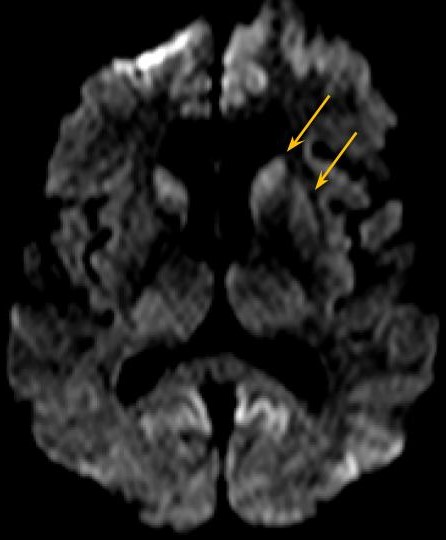
Fig. 4 - Sequência DWI, plano axial mais inferior, demonstrando restrição a difusão nos núcleos caudados e putâmens.
Artículo recibido en miércoles, 29 de abril de 2020
Artículo aceptado el martes, 12 de mayo de 2020
 Todos los artículos científicos publicados en brad.org.br están bajo una licencia Creative Commons.
Todos los artículos científicos publicados en brad.org.br están bajo una licencia Creative Commons.
 Portugués PDF
Portugués PDF
 Impresión
Impresión
 Enviar este artículo por correo electrónico
Enviar este artículo por correo electrónico
 Cómo citar este artículo
Cómo citar este artículo
 Enviar un comentario
Enviar un comentario
 Mendeley
Mendeley
 Pocket
Pocket