Cahinã Odilon Gobbo Da Silva 1, Fabio Hiroshi Okuyama 2, Rafael Salvajolli Ribeiro 3
Resumo
A doença da urina de xarope de bordo é uma rara doença hereditária. Trata-se da disfunção do complexo enzimático alfa-cetoácido-desidrogenase de cadeia ramificada, levando ao aumento da concentração dos aminoácidos de cadeia ramificada no plasma. Essa condição, sem o adequado tratamento, resulta em sequelas neurológicas irreversíveis. Assim, o reconhecimento do padrão radiológico da doença é fundamental para o precoce diagnóstico e o início do tratamento, prevenindo a deterioração neurológica.Dados do caso
Feminino, 0 anos.
Palavras chaves
Doença da Urina de Xarope de Bordo, Erros Inatos do Metabolismo dos Aminoácidos, Aminoácidos de Cadeia Ramificada, Imagem por Ressonância Magnética.
Histórico Clínico
Recém-nascido do sexo feminino, a termo, 2 dias de vida, evoluiu com quadro de hipoglicemia persistente. Realizado Ressonância Magnética de Crânio para investigação, a qual sugeriu o diagnóstico de doença da urina de xarope de bordo. Posteriormente, a cromatografia sérica de aminoácidos confirmou o diagnóstico.
Achados Radiológicos
As imagens da Ressonância Magnética de Crânio demonstram lesões hiperintensas em T2 (figuras 1 e 2) com restrição no estudo por difusão (figura 3), comprometendo o tronco cerebral na região do bulbo, ponte, mesencéfalo e os tálamos/braços posteriores das cápsulas internas, de forma simétrica e bilateralmente, sugerindo lesões de natureza neurometabólica.
Discussão
A doença da urina de xarope de bordo é uma rara doença metabólica hereditária de caráter autossômico recessivo causada pela disfunção da atividade do complexo alfa-cetoácido-desidrogenase de cadeia ramificada. A deficiência desse complexo enzimático resulta em níveis elevados de aminoácidos de cadeia ramificada (leucina, valina e isoleucina) no plasma, na urina e no líquido cefalorraquidiano [1]. A doença pode se manifestar de cinco formas: clássica, intermediária, intermitente, tiamina – responsível e deficiência de dihidrolipoamida desidrogenase (E3). A forma clássica é a mais prevalente e a que mais oferece riscos ao paciente, justamente por ser aquela com o maior déficit na atividade do complexo enzimático. Nessa apresentação da doença, os sintomas se manifestam entre o quarto e o sétimo dia após o nascimento. A criança inicia o quadro clínico com letargia, recusa alimentar ou sucção débil, perda de peso, grave cetoacidose, cheiro característico de açúcar queimado na urina e sinais neurológicos de intoxicação, podendo evoluir para o coma e óbito [1]. A elevada concentração de leucina é a principal responsável pela injúria encefálica, representada por típico edema tanto intramielínico quanto vasogênico [2]. O provável mecanismo para o edema intramielínico é o acúmulo de cetoácidos e de moléculas de água entre as camadas de mielina enquanto o mecanismo para o edema vasogênico é a ruptura da barreira hematoencefálica durante as crises de decompensação metabólica ocasionadas pela doença [2]. O padrão da injúria encefálica no estudo por Ressonância Magnética de Crânio consiste em áreas hiperintensas em T2, com restrição à difusão e hipointensas em T1, principalmente na substância branca do cerebelo, braço posterior da cápsula interna, tálamo, globo pálido e tronco encefálico. Na Tomografia Computadorizada de Crânio, as imagens podem mostrar hipodensidade simétrica bilateral acometendo substância branca do braço posterior da cápsula interna, tálamo, mesencéfalo e substância branca cerebelar [3,4]. Os achados radiológicos são sugestivos da doença, no entanto, deve-se ter a confirmação laboratorial para realizar o diagnóstico. Essa é feita pela detecção da elevação dos níveis séricos de leucina, valina e isoleucina, efetuada pela cromatografia sérica de aminoácidos [1]. O tratamento consiste na instituição de uma dieta alimentar adequada, hipoproteica, restrita em aminoácidos ramificados [1].
Lista de Diferenciais
Diagnóstico
Aprendizado
O reconhecimento do padrão radiológico da doença da urina de xarope de bordo é essencial para auxiliar no diagnóstico precoce da doença, o qual é fundamental na prevenção da deterioração neurológica que ocorre na ausência do tratamento nutricional adequado.
Referências
1. Blackburn PR, Gass JM, Vairo FP, Farnham KM, Atwal HK, Macklin S et al. Maple syrup urine disease: mechanisms and management. Appl Clin Genet. 2017; 10: 57-66.
2. Reddy N, Calloni SF, Vernon HJ, Boltshauser E, Huisman TAGM, Soares BP. Neuroimaging Findings of Organic Acidemias and Aminoacidopathies. (2018) Radiographics. 38 (3): 912-931.
3. Jain A, Jagdeesh K, Mane R, Saurabh S. Imaging in classic form of maple syrup urine disease: a rare metabolic central nervous system. J Clin Neonatol. 2013; 2 (2): 98-100.
4. Xia W, Yang W. Diffusion-weighted magnetic resonance imaging in a case of severe classic maple syrup urine disease. J. Pediatr. Endocrinol. Metab. 2015; 28: 7-8.
Imagens
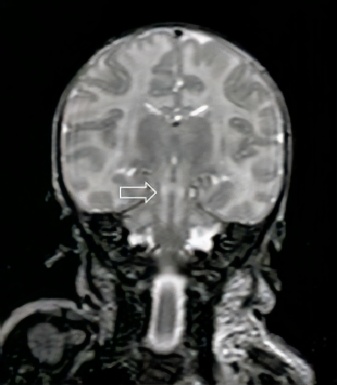
Figura 1: Imagens de RM em T2 demonstram hipersinal no tronco encefálico e braços posteriores da cápsula interna.
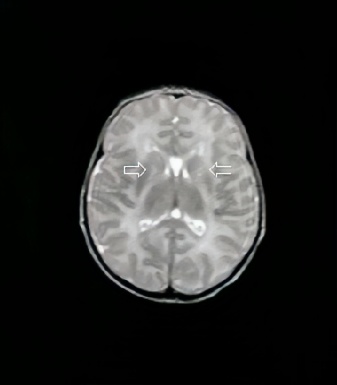
Figura 2: Imagens de RM em T2 demonstram hipersinal no tronco encefálico e braços posteriores da cápsula interna.
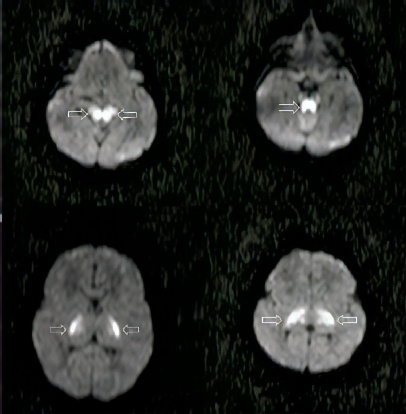
Figura 3: Imagens de RM demonstram restrição no estudo por difusão no mesencéfalo, ponte e tálamos.
Artigo recebido em terça-feira, 14 de abril de 2020
Artigo aprovado em quarta-feira, 29 de abril de 2020
 Todos os artigos científicos publicados em brad.org.br são licenciados sob uma licença Creative Commons.
Todos os artigos científicos publicados em brad.org.br são licenciados sob uma licença Creative Commons.
 Português PDF
Português PDF
 Imprimir
Imprimir
 Envie este artigo por e-mail
Envie este artigo por e-mail
 Como citar esse artigo
Como citar esse artigo
 Envie um comentário
Envie um comentário
 Mendeley
Mendeley
 Pocket
Pocket